Splenectomy for the Management of Primary Myelofibrosis.
Σπληνεκτομή για την αντιμετώπιση Πρωτοπαθούς Μυελοίνωσης.
Abstract
Primary myelofibrosis is a myeloproliferative neoplasm, which is characterized by myeloproliferation of clones arising from stem cells, which frequently but not always exhibit JAK2, CALR, or MPL mutations. Other findings include bone marrow fibrosis, abnormal expression of inflammatory cytokines, anemia, hepatosplenomegaly, extramedullary hematopoiesis, constitutional symptoms, cachexia, risk of progression to leukemia, and shortened survival.
Affected patients frequently present debilitating symptoms, including intense pain and the sensation of early satiety, in addition to cellular sequestration/withdrawal resulting in severe cytopenia. JAK2 inhibitors, such as ruxolitinib and fedratinib, constitute the mainstay of treatments as they cause significant and durable reduction of splenic volume. Unfortunately, many patients who do not fall within the indications for treatment with a JAK2 inhibitor become resistant to treatment over time. Fortunately, new therapies are being developed which reduce the degree of splenomegaly in some patients. Nevertheless, splenectomy, splenic irradiation, and partial embolization of branches of the splenic artery remain valuable therapeutic options in selected patients.
Introduction
Philadelphia chromosome-negative myeloproliferative neoplasms include essential thrombocythemia, polycythemia vera, and myelofibrosis. These morbid conditions are clinically characterized by systemic symptoms, a predisposition to thrombosis, and varying degrees of splenomegaly.Although relatively rare in essential thrombocythemia, splenomegaly is found in approximately one-third of patients with polycythemia vera and much more frequently in myelofibrosis. The development of marked splenomegaly in patients with myelofibrosis results in 35% of them having a palpable spleen at least 10 cm below the left costal margin, while in 23% of patients the spleen extends more than 16 cm below the costal margin.
Symptoms resulting from splenomegaly include pressure and pain in the left upper quadrant of the abdomen, left scapular pain, and early satiety due to pressure on the stomach.Additionally, massive splenomegaly may lead to the development of portal hypertension and its consequences, due to the high pressures transmitted to the portal system. Patients with myelofibrosis may develop splanchnic vein thrombosis, extramedullary hepatic hematopoiesis, and obstructive portal venopathy.Excessive splenomegaly may cause compression of the inferior vena cava and iliac veins, resulting in lower extremity edema. In some cases, the extent of splenomegaly may lead to the development of ischemic areas, resulting in painful splenic infarcts.The detrimental impact of splenomegaly on the quality of life of patients with myelofibrosis has been documented in many studies, from which it is readily apparent that reduction of spleen size constitutes one of the 5 main therapeutic goals. The presence and degree of splenomegaly negatively affects the prognosis of patients with polycythemia vera and essential thrombocythemia.
Assessment of splenomegaly is performed by palpation in the left upper quadrant of the abdomen, using the left costal margin as a reference point. If the spleen is not palpable, the patient is placed in the right lateral decubitus position to bring the organ into a more favorable position for palpation.If palpation is still not possible, imaging assessment is required using ultrasonography, CT, or MRI. The mean splenic volume in healthy adults is 166 cm3, and the mean length is 10.9 cm. Typically, a spleen length greater than 12 cm on imaging is considered pathological, although factors such as height and ethnicity must be taken into account.
The predominant pharmacological treatment for reducing spleen size in myelofibrosis is the use of JAK2 inhibitors, such as ruxolitinib and fedratinib.These molecular inhibitors have proven very effective in improving splenomegaly and have fundamentally changed the approach to splenomegaly in myelofibrosis. However, approximately 15% of patients are unable to receive JAK2 inhibitor therapy, while many develop resistance to treatment.
Various studies report high rates of mandatory discontinuation of ruxolitinib therapy, at 50% of patients at 3 years and 72.8% at 5 years. Additionally, patients treated with ruxolitinib have a median survival of only 14 months; therefore, investigation of other avenues for managing splenomegaly in patients with myelofibrosis is required.
ANATOMY AND PATHOPHYSIOLOGY.
The spleen is organized into two distinct compartments: the white and red pulp. The white pulp serves as a reservoir for lymphocytes, while the red pulp functions by filtering the blood. The red pulp is organized into a loose network of capillaries and sinuses.Littoral cells (large phagocytic endothelial cells) line the sinus walls and act as red blood cell filters, while splenic vascular endothelial cells line the capillary walls. These cells have distinct cellular functions, with littoral cells functioning as cellular filters and waste collectors.The spleen is covered by a capsule of dense fibrous tissue and smooth muscle fibers. Blood enters through the splenic artery, which branches into smaller arterioles. Approximately 90% of total splenic blood flows through the venous sinuses, bypassing the red pulp filtration system.Blood exits through the splenic vein, which joins the superior mesenteric vein to form the portal vein.
Splenomegaly in myeloproliferative neoplasms is predominantly due to extramedullary hematopoiesis. The abnormal circulation of hematopoietic stem cells, due to the dysregulated bone marrow microenvironment, leads to their implantation in extramedullary sites such as the spleen.
CONVENTIONAL CYTOREDUCTIVE THERAPIES.
For the management of symptomatic splenomegaly, there is a considerable number of therapies with proven effectiveness. Before the advent of JAK2 inhibitors, various cytoreductive agents were used to reduce splenomegaly in myelofibrosis.Hydroxyurea, a ribonucleotide reductase inhibitor, was used in patients with hyperproliferative findings, including splenomegaly, leukocytosis, and constitutional symptoms, who could not receive JAK2 inhibitor therapy.Administration of low-dose melphalan is moderately effective in reducing splenomegaly but carries the risk of cytopenia and predisposition to leukemic transformation. The immunomodulatory agents thalidomide and lenalidomide have been widely used in myelofibrosis, and although they demonstrated ability to improve anemia, they contributed little (10–20%) to the reduction of splenomegaly.
JAK 2 INHIBITORS.
The most effective pharmacological tool for managing splenomegaly in myelofibrosis is JAK2 inhibition. The mechanism of action is not fully elucidated but may be related to the inhibition of JAK 2-mediated hematopoietic stem cell migration through the CXCL12/CXCR4 pathway.Despite the initial enthusiasm with JAK 2 inhibitors, it must be emphasized that patients unfortunately develop tolerance to their use, and simultaneously, these drugs have no effect on the natural progression of myelofibrosis.
SPLENECTOMY.
Before the introduction of JAK2 inhibitor therapy, splenectomy was a common option for relief of splenomegaly-related symptoms. Additionally, the procedure helped improve anemia and thrombocytopenia, allowing the postoperative release of sequestered cells into the peripheral circulation.
Many clinicians advocate performing splenectomy before allogeneic transplantation to improve engraftment. However, the advantages of this strategy remain a subject of debate.
Today, splenectomy constitutes a valuable therapeutic strategy, particularly in patients who are ineligible for, develop intolerance to, or do not respond to JAK inhibitor therapy and require symptomatic treatment.However, careful patient selection is the key to success, and it is important to attempt re-administration of JAK2 inhibitors postoperatively to prevent, as much as possible, accelerated hepatomegaly due to extramedullary hematopoiesis in the liver.
Concerns regarding acceleration of leukemic transformation in patients with myelofibrosis after splenectomy have been raised by many over time. Regardless, the palliative role of splenectomy in properly selected patients cannot be underestimated.Today, splenectomies should be performed in centers with experience and expertise, always considering the complex nature of this operation in patients with myelofibrosis.
Before splenectomy, patients must be vaccinated against 1) Streptococcus pneumoniae, 2) Haemophilus influenzae, and 3) Neisseria meningitidis, as asplenia increases the risk of life-threatening infections from encapsulated organisms.Ideally, vaccinations should begin 10 weeks before splenectomy, although an adequate immune response occurs after 2 weeks if emergency splenectomy is required.
SPLENIC IRRADIATION.
In patients who cannot undergo surgery due to poor general condition, splenic irradiation may be an option.The spleen is very radiosensitive, and therefore even a small dose may be effective. Additionally, patients with ascites due to extramedullary hematopoiesis may be managed with radiation therapy. The most commonly administered dose is 10 Gy in 10 fractions.However, it must be emphasized that splenic irradiation is accompanied by many complications. The most significant is bone marrow suppression, which results in worsening of disease-related anemia and thrombocytopenia and the development of neutropenia.Another important manifestation of toxicity from the gastrointestinal tract is the development of nausea and diarrhea, which further complicates the clinical picture. Finally, for patients who have exhausted the option of JAK2 inhibitor therapy and cannot tolerate splenectomy, splenic irradiation constitutes a potentially therapeutic option for managing symptomatic splenomegaly.
PARTIAL SPLENIC ARTERY EMBOLIZATION.
Splenic artery embolization for the management of massive splenomegaly was first used as a non-invasive method in 1970. However, this procedure was accompanied by significant complications such as splenic rupture, splenic abscess, sepsis, and death.Therefore, partial splenic artery embolization was proposed instead, incorporating antibiotic prophylaxis, with a reduced likelihood of complications. Furthermore, a portion of splenic tissue is preserved, which can maintain splenic function.This technique was initially applied in cirrhotic patients who were poor surgical candidates, and later it was also used in cases of splenic trauma to control hemorrhage. In practice, the major limitation for the application of partial splenic artery embolization is the development of post-embolization syndrome, characterized by severe pain in the left upper quadrant, requiring hospital admission, hydration, and administration of strong analgesics.This complication is encountered in 75% of cirrhotic patients who underwent partial splenic artery embolization. Other complications such as splenic abscess, portal vein thrombosis, pleural effusion, and worsening of ascites may occur.
Case Report
A 65-year-old female patient suffering from chronic idiopathic myelofibrosis for the past 10 years developed massive splenomegaly. The consequences of this mass included the development of severe abdominal pain, compression of the inferior vena cava, lower extremity edema, and dyspnea.Her case was refractory to various pharmacological treatments, including JAK2 inhibitors, and therefore splenectomy was recommended by the hematologists to improve her quality of life.
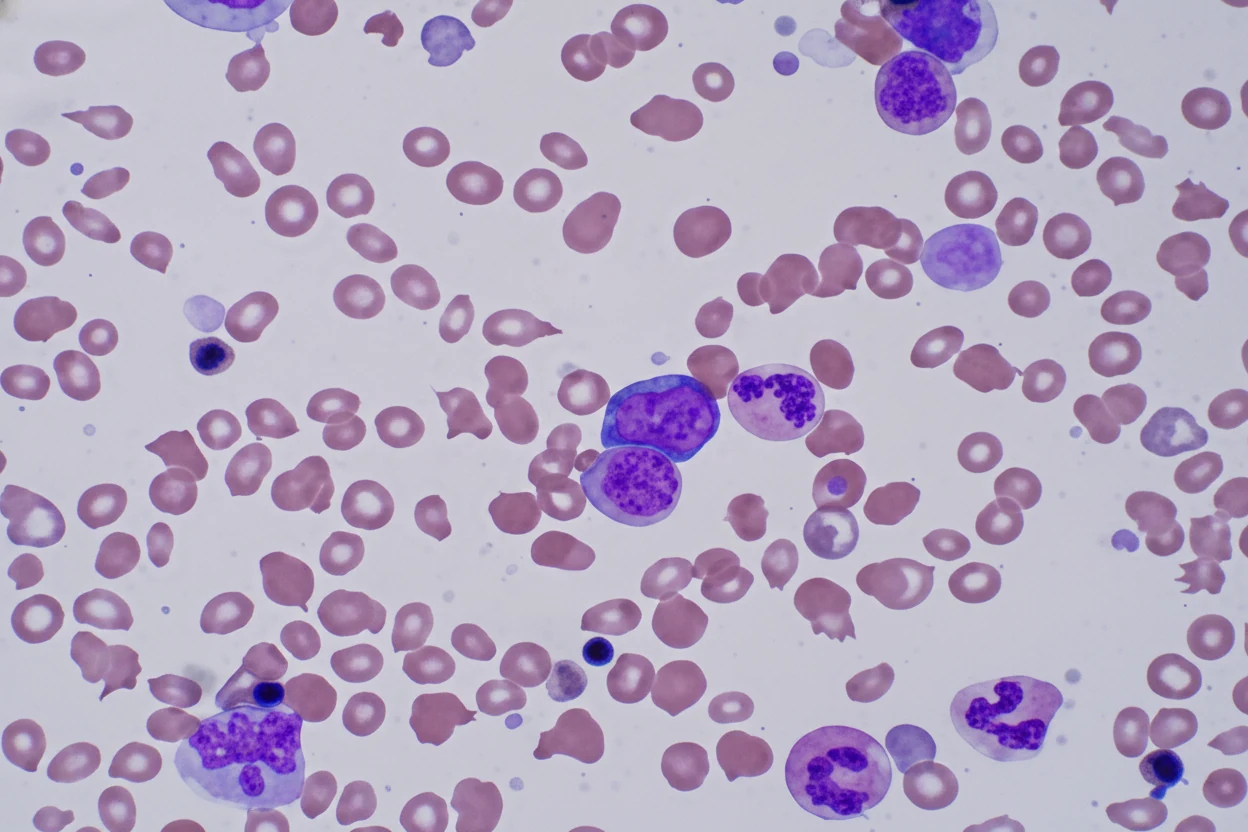
At admission, she was anemic (hemoglobin 6.8 g/dL), thrombocytopenic (platelets 92 x 10^3/L), with marked leukocytosis (64.0 x 10^3/L).On peripheral blood smear examination, abundant circulating nucleated red blood cells, poikilocytosis, anisocytosis, immature granulocytes, and giant platelets were found.
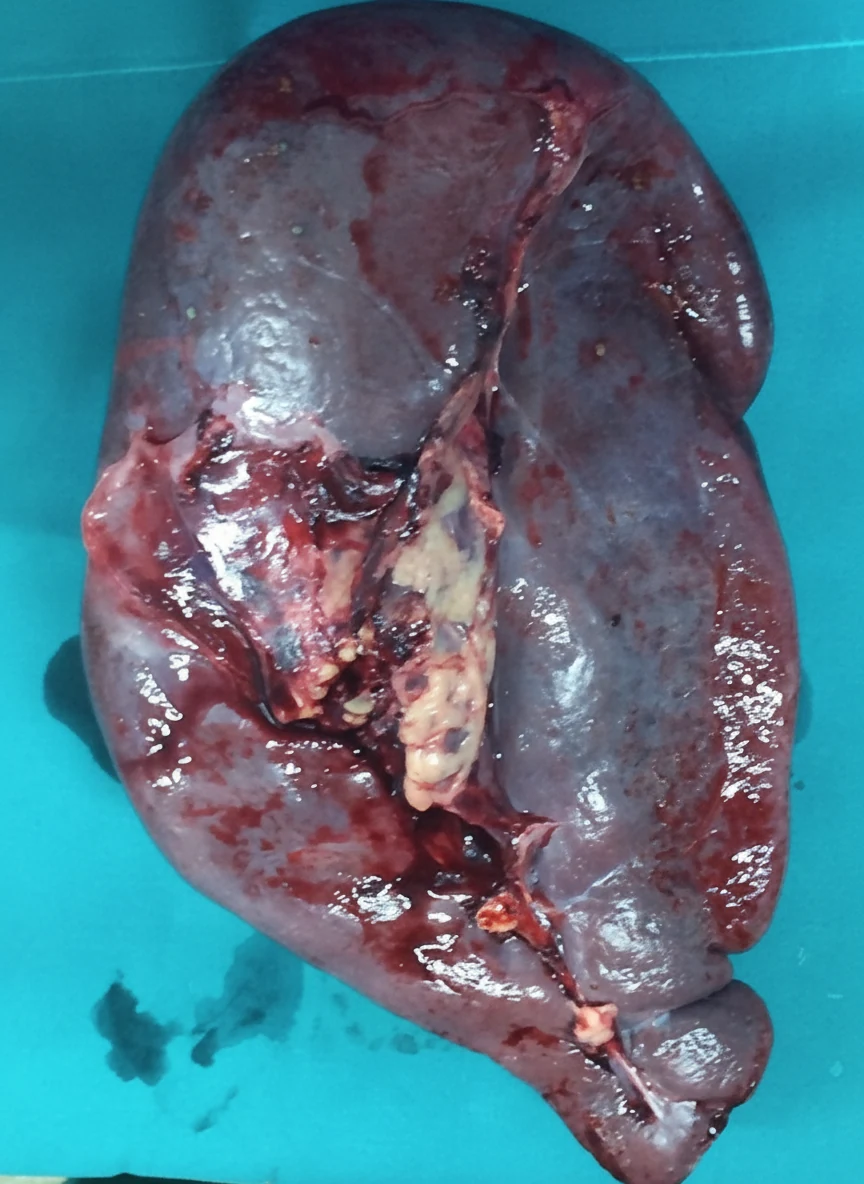
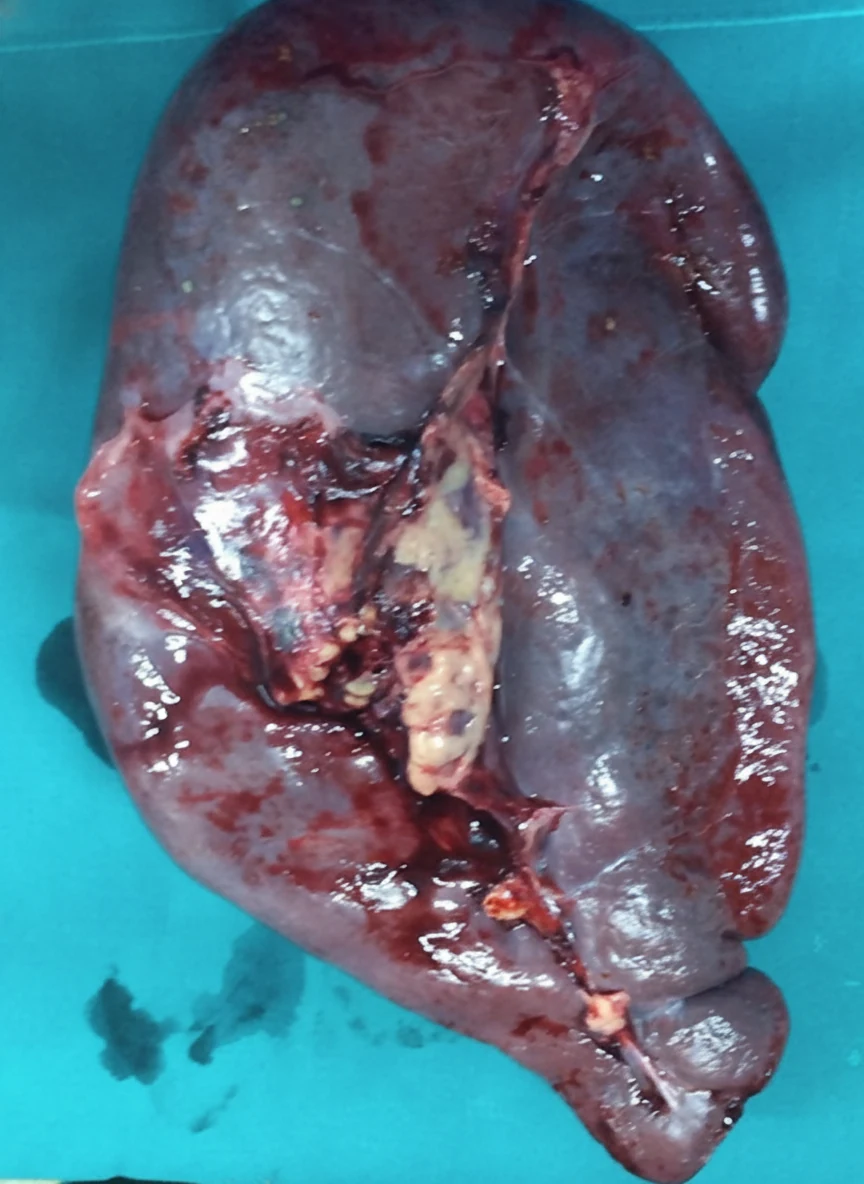
Through a left Kocher incision, which was extended to the right, the patient underwent splenectomy and cholecystectomy for the management of concomitant cholelithiasis.The weight of the spleen was 3.9 kg. Additionally, a liver biopsy was obtained, which revealed the presence of extramedullary hematopoiesis.
The patient's postoperative course was uneventful, and she was discharged 8 days later. At discharge, her anemia had improved (hemoglobin 9.2 g/dL), platelets were 99 x 10^3/L, while white blood cells were unchanged (64.0 x 10^3/L), and numerous nucleated red blood cells continued to be found on peripheral blood smear examination.
Three months later, there was clear improvement in the patient's clinical condition, but the hematological picture remained unchanged. This most likely reflected active extramedullary hematopoiesis in the liver, in an effort to compensate for the fibrotic bone marrow.
Conclusions and Recommendations
Splenectomy is frequently performed to manage symptomatic splenomegaly that is refractory to pharmacological treatment. Indications for splenectomy in patients with myelofibrosis include: 1) abdominal pain and discomfort, 2) symptomatic portal hypertension, 3) severe thrombocytopenia, and 4) need for frequent blood transfusions.The effectiveness of the procedure has been evaluated in numerous studies, particularly highlighting improvement in anemia, thrombocytopenia, and portal hypertension, as well as elimination of splenomegaly-related symptoms. However, splenectomy in patients with myelofibrosis is associated with numerous complications, such as reactive thrombocytosis during the postoperative period, thrombo-hemorrhagic events, significant perioperative morbidity and mortality, and disease transformation.
Complications following splenectomy are classified as perioperative and late. Among perioperative complications, which are observed in approximately 30% of patients, the most frequently reported are hemorrhage (14%), thrombosis (13%), and infections (9%). The risk of thrombosis can be reduced with prophylactic administration of low-molecular-weight heparins postoperatively.These complications can prove fatal in 8% of splenectomized patients, with the leading causes of death being infections and hemorrhage. Advanced age, thrombocytopenia (50–100 x 10^9/L), circulating blasts, leukocytosis (>25 x 10^9/L), and a hypocellular bone marrow constitute risk factors for adverse outcome following splenectomy.
Symptomatic splenomegaly constitutes a scientific challenge in the effort to provide relief for patients with myelofibrosis. Despite the advent of JAK2 inhibitors, many patients develop intolerance or eventually develop resistance to these drugs.As a consequence, new pharmacological therapies are being developed; however, the strategies of splenectomy, splenic irradiation, and partial arterial embolization of the organ continue to hold their place in the armamentarium of physicians for managing splenomegaly in patients with myelofibrosis.
Patients who meet the indications for JAK2 inhibitor use (e.g., adequate platelet count) should initially be treated with one of the available agents, ruxolitinib or fedratinib. Patients who do not meet the criteria or in whom JAK2 inhibitor therapy has failed (use of one or more agents) should be evaluated for enrollment in clinical trials of new drugs.
If they still do not meet the criteria for clinical trial enrollment, splenectomy constitutes the first option, taking into account both the effectiveness and safety of the procedure. The benefits of splenectomy must always be weighed against the immediate and long-term risks, complications, and morbidity.Although splenectomy is not routinely performed before transplantation, it may be the appropriate management in patients with massive splenomegaly and related symptoms, as in these cases there is an increased risk of graft rejection.It is very important that splenectomy be performed with strict criteria in specially selected patients (e.g., those who present with severe refractory cytopenia and extremely large symptomatic splenomegaly who have clearly failed to respond to pharmacological therapy).The recent approval of JAK1/2 inhibitors in patients with myelofibrosis has diminished the role of splenectomy.
In patients who do not meet the criteria for surgical intervention (poor general condition, comorbidities), splenic irradiation and partial splenic artery embolization are considered alternative approaches.These approaches are always dependent on the department's policy, the patient's wishes, and the balance of expectations/outcomes and, of course, the anticipated toxicity. Splenic irradiation may reduce spleen size, but the effect is short-lived, while the risk of severe and prolonged cytopenia constitutes a deterrent factor for its application.
In conclusion, the modern approach to managing splenomegaly in patients with myelofibrosis has fundamentally changed over the past decade and continues to evolve rapidly. Pharmacological options are expanding, including agents beyond JAK2 inhibitors.Nevertheless, for many patients, splenectomy, splenic irradiation, and partial splenic artery embolization remain valuable second-line options. Current efforts are focused on the development of new therapeutic agents that could potentially eliminate malignant hematopoietic stem cells and thereby restore normal hematopoiesis and eliminate extramedullary hematopoiesis, which results in splenomegaly, and ultimately increase life expectancy.
- Splenectomy for PMF is reserved for refractory symptomatic splenomegaly after failed medical therapy
- Operative mortality is 5-10% due to portal hypertension and coagulopathy
- Portal vein thrombosis occurs in 8-20% postoperatively requiring anticoagulation
- Hepatic extramedullary hematopoiesis may progress after splenectomy
- JAK2 V617F mutation is present in ~60% of PMF cases; ruxolitinib is the current medical standard
Sign in to join the discussion
Loading...