Primary Retroperitoneal Angiosarcoma.
Πρωτοπαθές Οπισθοπεριτοναικό Αγγειοσάρκωμα.
Abstract
Primary retroperitoneal sarcomas are rare mesenchymal tumors, with liposarcoma being most common and angiosarcoma an exceptionally uncommon and highly aggressive variant with poor prognosis. We report a 68-year-old patient admitted to a regional hospital with acute abdominal pain, respiratory distress, and hypotension. After resuscitation, abdominal CT revealed a large (10 x 6.8 cm; craniocaudal 16.4 cm) multilobulated, low-density retroperitoneal mass in the left iliac fossa, extending from the lower pole of the left kidney to the coccyx, posterior to the external iliac vessels. Thoracic staging was negative. Radical surgical resection was performed, and histopathology with immunohistochemistry confirmed primary retroperitoneal angiosarcoma. Adjuvant therapy was planned in the multidisciplinary setting. This case underscores the aggressive behavior of retroperitoneal angiosarcoma and emphasizes the importance of early diagnosis and radical surgical resection for any chance of improved survival.
Introduction
Primary retroperitoneal sarcoma is a malignant tumor arising from the mesenchymal cells of the retroperitoneal space. These tumors are rare, with an incidence of 0.0027%. Among the various types, liposarcoma is the most common, while malignant peripheral nerve sheath tumors and angiosarcomas are encountered more rarely.
Angiosarcoma exhibits a high degree of malignancy and therefore its prognosis is poor. Early diagnosis and appropriate therapeutic management have an impact on the survival of these patients.
The present report describes a primary retroperitoneal sarcoma in a middle-aged patient.
Case Report
A 68-year-old patient was admitted to a regional hospital, complaining of acute abdominal pain, respiratory distress, and a drop in blood pressure. After initial resuscitation, an abdominal CT scan was performed, which revealed the presence of a large, multilobulated, low-density soft-tissue lesion in the region of the left iliac fossa, with indicative dimensions of 12 x 6.9 cm.The lesion extended superiorly up to the lower pole of the left kidney, and descended posterior to the external iliac vessels down to the level of the coccygeal vertebrae (indicative craniocaudal diameter 16.6 cm). The remaining abdominal organs were normal, as was the subsequently performed CT scan of the thorax, with no evidence of metastatic foci in the liver or lungs.
After stabilization, the patient reported that he had been experiencing a dull pain in the left lumbar region for the past 2 months, which he attributed to constipation. His personal and family medical history was unremarkable. On clinical examination, marked abdominal distension was noted, with guarding on the left side without peritonism, and diminished bowel sounds.No masses were palpable. Hemoglobin was 8 g/dL, and two units of packed red blood cells were transfused. Prothrombin time was normal, as were liver function tests and other biochemical studies. Catecholamine, cortisol, and aldosterone levels were within normal limits.The following day, at the family's request, the patient was transferred to our clinic for further management.
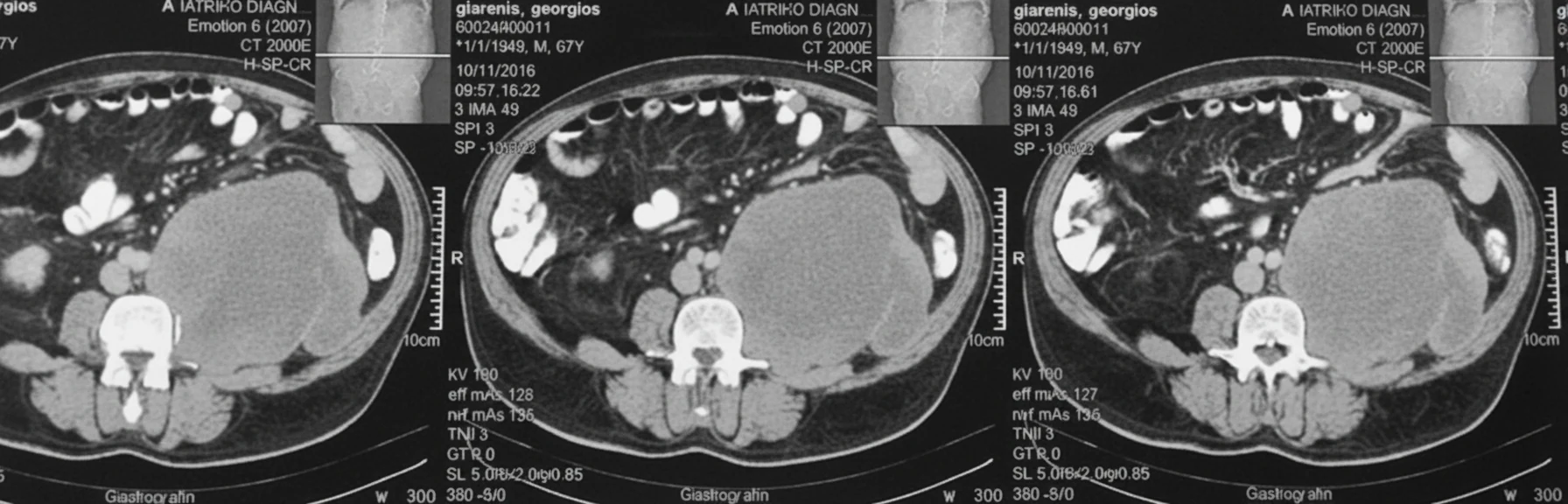
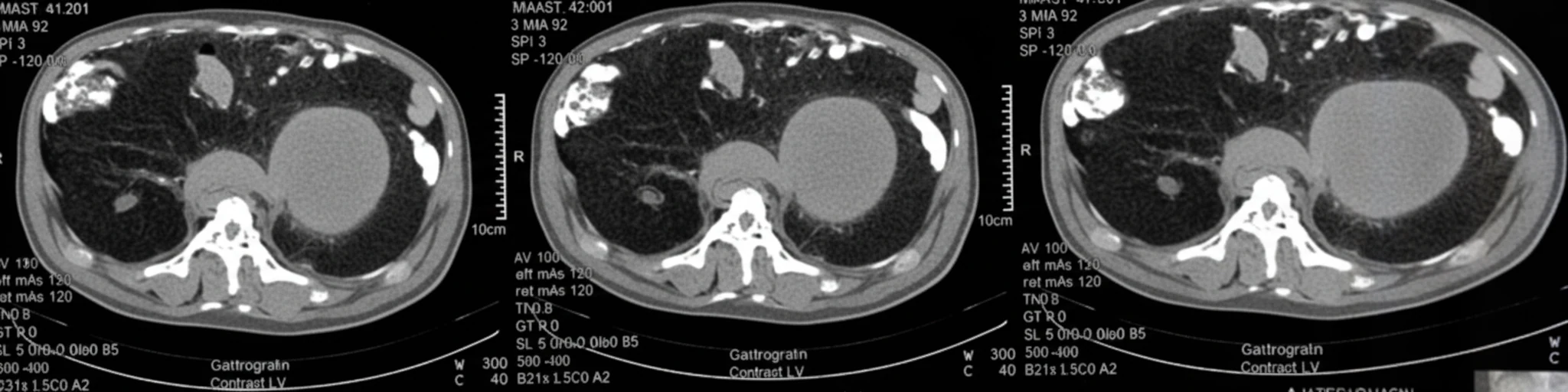
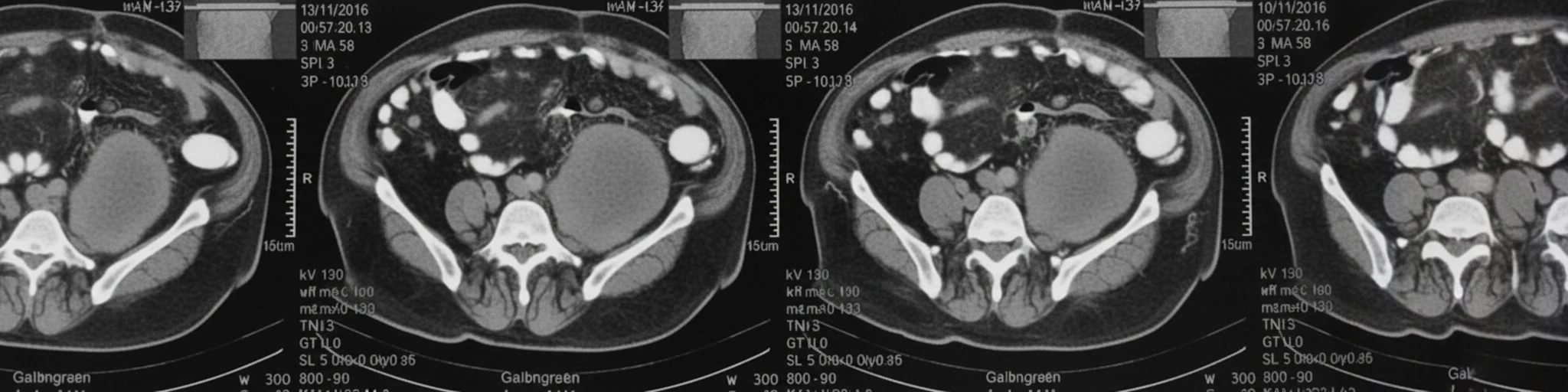
Taking into account the patient's unremarkable medical history, the presence of a resectable retroperitoneal tumor that had hemorrhaged (drop in BP and Hb), the absence of encasement/infiltration of critical vessels by the tumor, the absence of metastases, the impending obstruction of the left hemicolon, and the need for a reliable diagnosis, we decided to proceed with exploratory laparotomy.
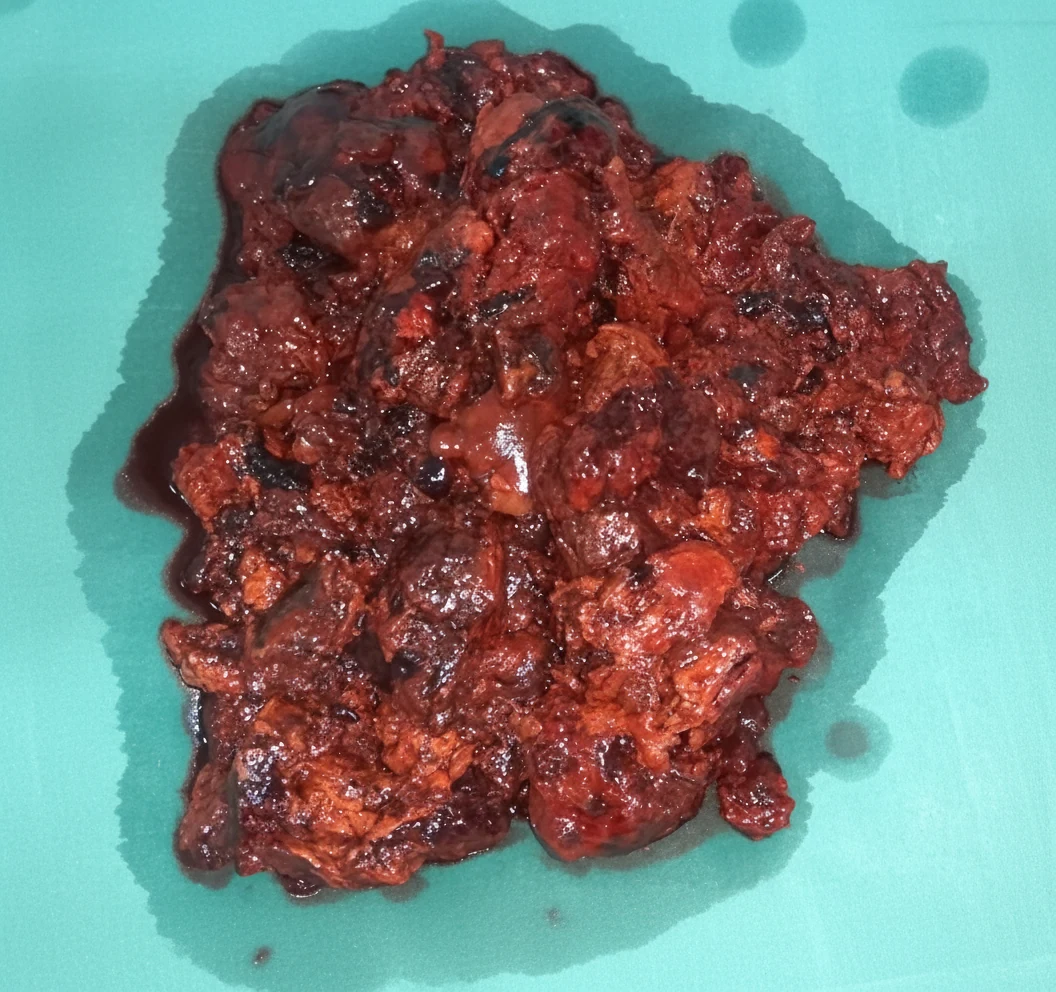
After opening the abdomen, we found hemoperitoneum, evidently caused by rupture and hemorrhage of the highly vascular retroperitoneal tumor. After evacuation of approximately 1 liter of blood and clots, a friable, dark-colored mass measuring 13 x 7.4 cm was exposed below the lower pole of the left kidney, with indistinct borders, firmly adherent to the left psoas muscle.The regional vascular structures were slightly distended due to compression by the mass, but no fluctuations in arterial pressure were observed during manipulation of the tumor. With careful maneuvers, and despite the patient's hemorrhagic diathesis, complete excision of the tumor was achieved without major bleeding, including the portions posterior to the external iliac vessels, en bloc with the descending colon and sigmoid, which were considered infiltrated by the tumor by contiguity - Hartmann's procedure.

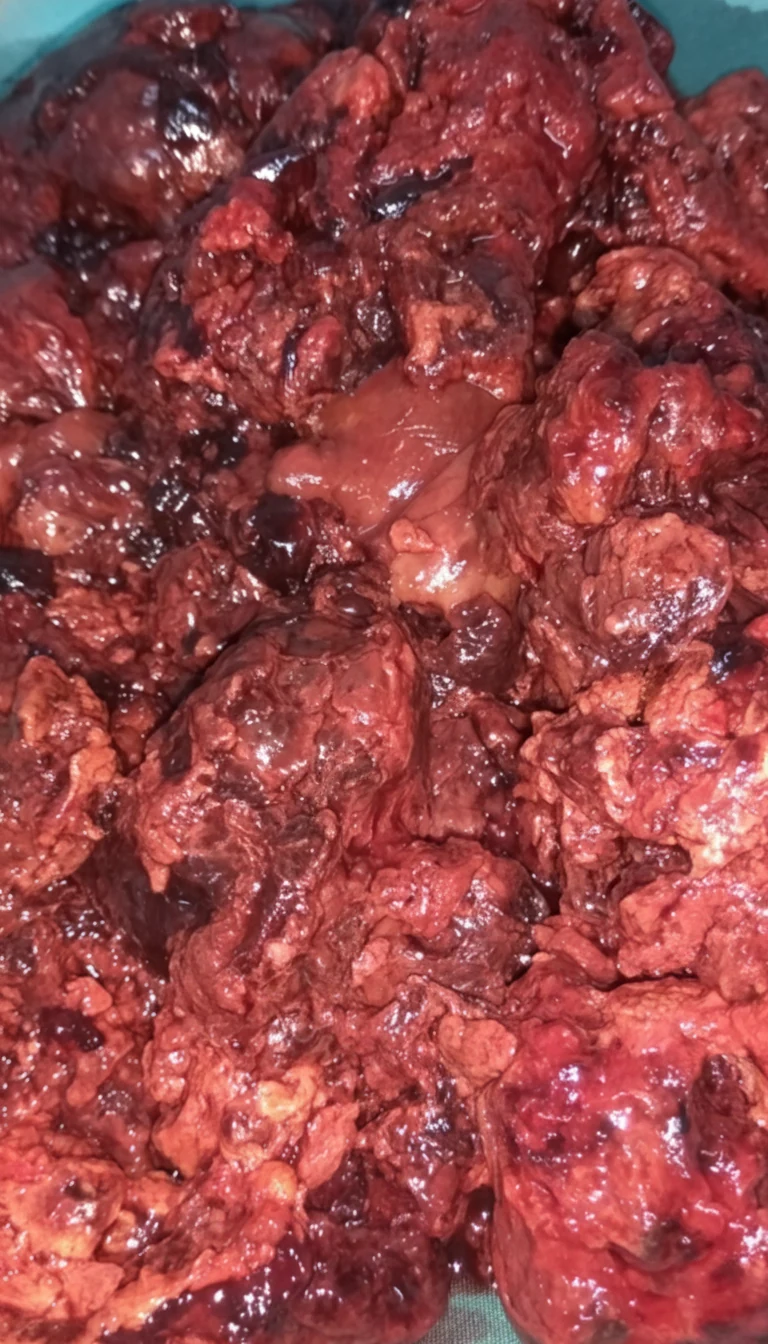
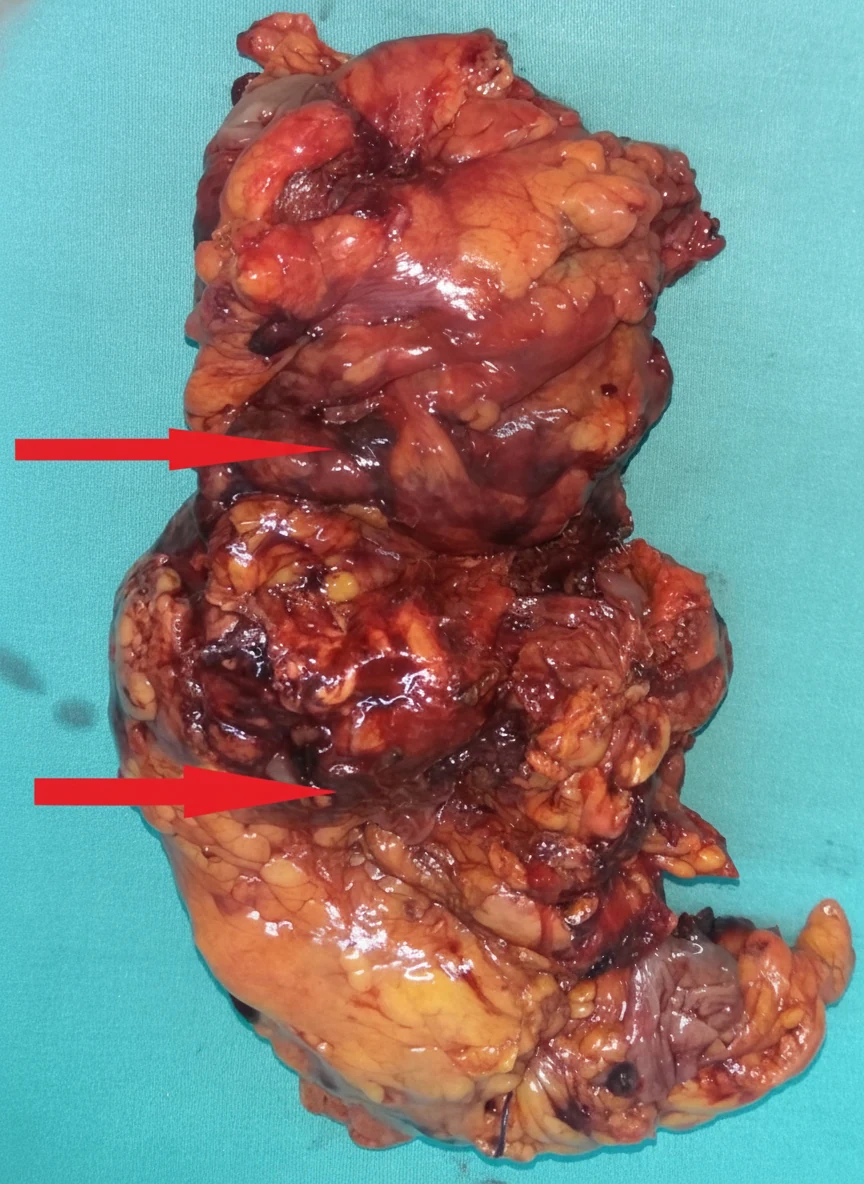
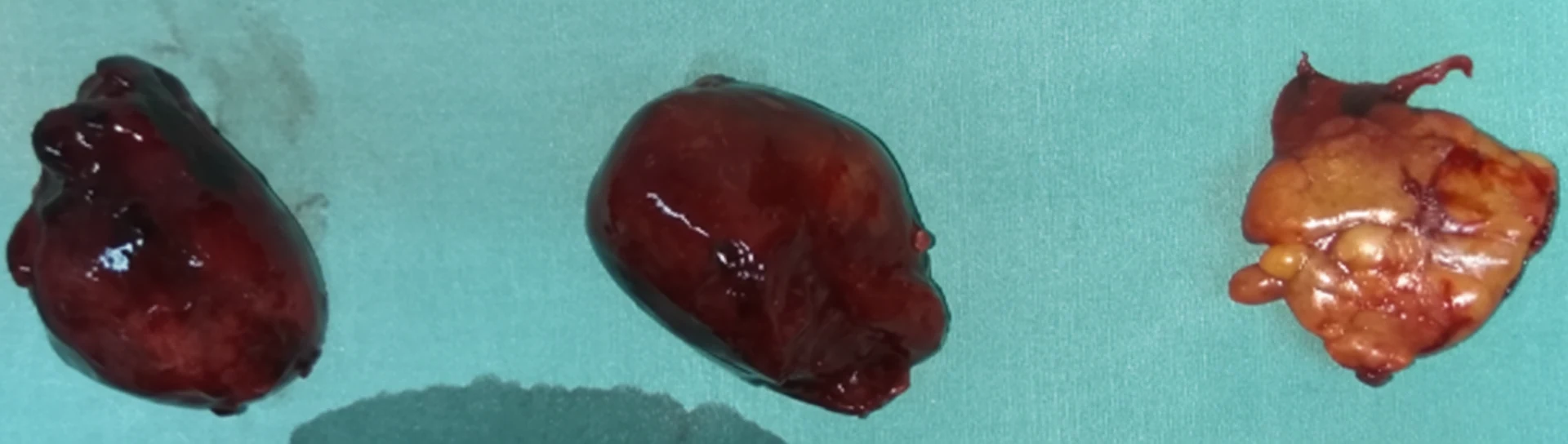
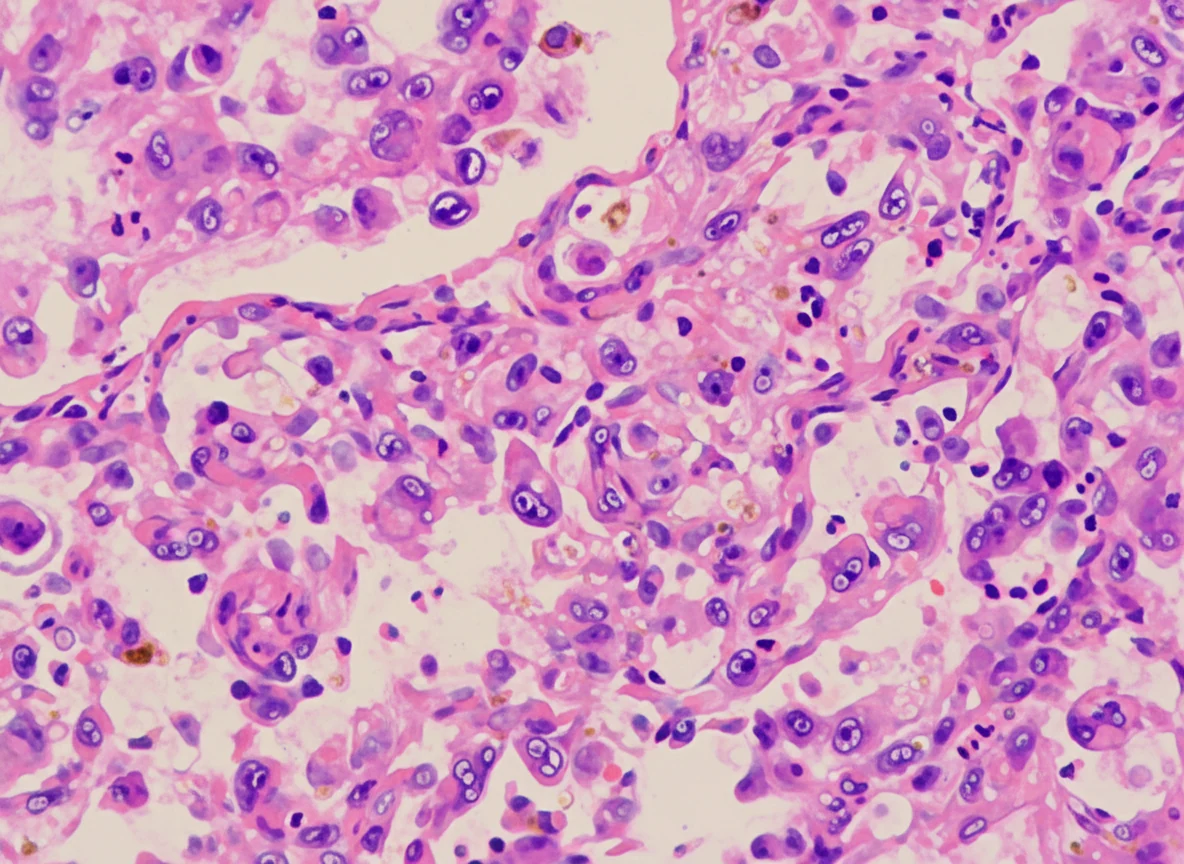
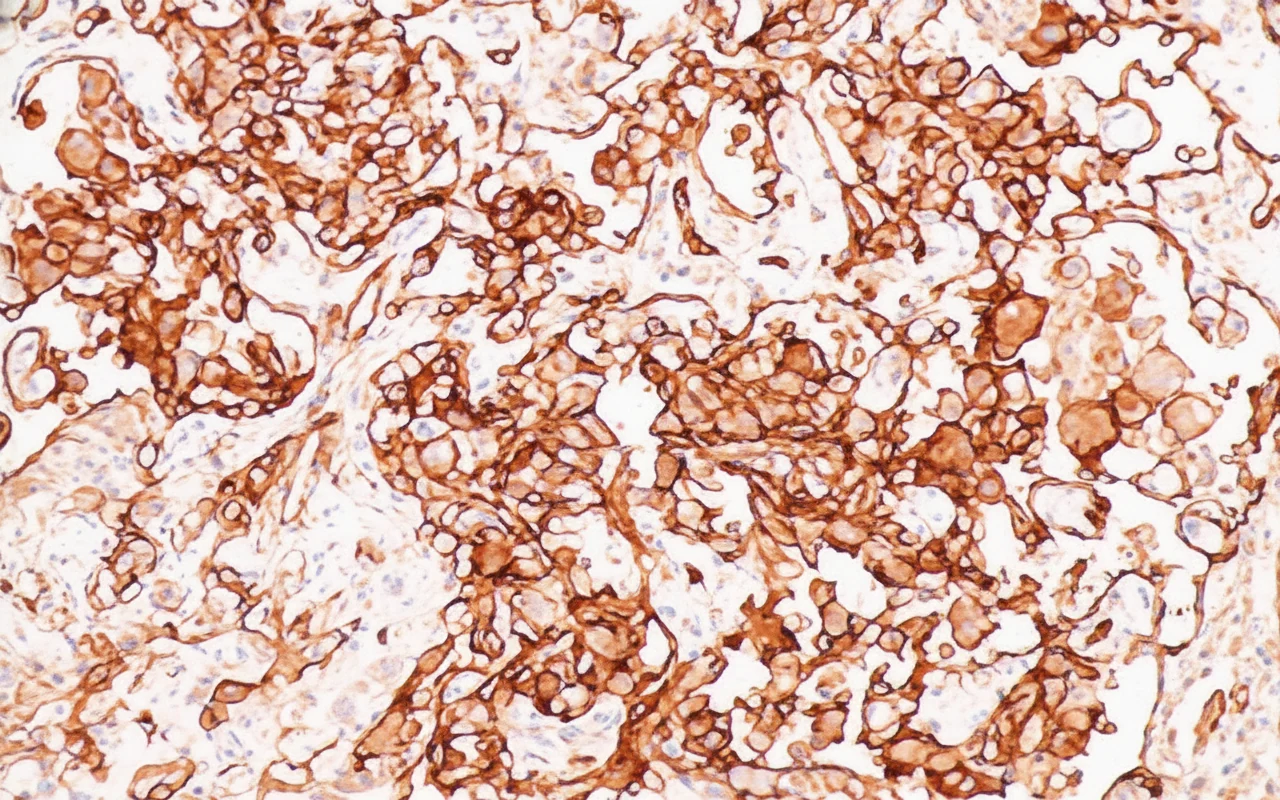
Histopathological examination revealed that the retroperitoneal mass was a malignant tumor with tissue necrosis within the neoplasm. The malignant cells were epithelioid cells with evident cellular atypia and intense mitotic activity. Immunohistochemistry demonstrated positive reactivity for pancytokeratin, vimentin, and CD31.In contrast, there was no reactivity to various markers such as epithelial membrane antigen, S100, transcription factor SOX-10, HMB-45, melan-A, podoplanin, CD117, thyroid transcription factor-1, chromogranin A, and smooth muscle actin.Finally, the Ki-67 proliferation index was approximately 50%.
The patient had an uneventful postoperative course and was discharged from our hospital 10 days later. Prior to discharge, he was evaluated by an oncologist and scheduled to receive adjuvant therapy. He remained symptom-free with good quality of life for a long period.
He died 18 months later, following the appearance of multiple pulmonary metastases.
Discussion
Angiosarcoma is an aggressive malignant tumor of endothelial cells arising from lymphatic or blood vessels, estimated to account for approximately 1% of all soft tissue sarcomas.
The pathological manifestations of angiosarcoma vary according to the grade of malignancy of the different tumors.For example, a low-grade angiosarcoma often presents as a small, solid focus/lesion with a small cell population and abundant transecting vessels. In contrast, the presentation of a high-grade angiosarcoma often includes hypercellularity, high mitotic activity, and atypical cells.Angiosarcoma can occur in any part of the body, at all ages, although it is more frequently encountered in the elderly, with a mean age of 60-71 years. It is most commonly found in the skin (50% of cases), whereas retroperitoneal localization accounts for 10-15% of cases.
The etiology of angiosarcoma has not been elucidated. Risk factors are considered to be exposure to radiation or radiotherapy, chronic lymphedema, and exposure to chemicals such as vinyl chloride. The patient in our case did not meet any of these factors.Recent studies demonstrate that radiation plays a significant role in tyrosine kinase receptors, particularly in the upregulation of MYC, KIT, and RET, as well as the downregulation of cyclin-dependent kinase inhibitor 2C.
The clinical manifestations of angiosarcoma depend mainly on its site of development. Superficial tumors often appear as rapidly growing soft tissue masses. In contrast, deeply located angiosarcomas are extremely difficult to identify early, except after symptoms develop due to pressure and/or pain.If left untreated, angiosarcomas grow rapidly, reaching 20 cm or even larger, with a risk of rupture and potentially massive hemorrhage, as in our case. The frequency of recurrence or metastasis is high, and therefore postoperative follow-up at regular intervals with imaging methods is required.The patient of our report died 18 months after surgical intervention, following the appearance of multiple metastases in both lungs. This confirms the concept that angiosarcoma is an aggressive malignancy with, unfortunately, a very poor prognosis.
The reported case supports the view of many studies that tumors larger than 5 cm are associated with a worse prognosis. Imaging examinations lack specificity for the diagnosis of angiosarcoma. Therefore, combined use of CT, MRI, and PET scan is required to determine the anatomy of the tumor and the extent of its spread.
CT examination of angiosarcoma reveals the following: 1) ovoid soft tissue masses, with or without a lobulated contour; 2) indistinct contours with densities lower than those of muscle; and 3) possible presence of hemorrhage, necrosis, or calcifications within the neoplastic mass.
MRI examination of angiosarcoma reveals the following: 1) T1-weighted images (T1WI) produce an inhomogeneous low signal; 2) T2-weighted images (T2WI) produce high-signal images.
Nevertheless, the gold standard for the diagnosis of angiosarcoma is immunohistochemistry.Considering that image-guided percutaneous biopsy causes minimal damage, carries a low risk of malignant cell seeding through the needle, and can provide sufficient diagnostic information, it constitutes the most effective method of histological diagnosis for this disease.Due to the particularities of its location, the retroperitoneal approach is the most preferred route for accessing this malignancy. If approach via this route is not feasible, the transperitoneal approach is followed as an alternative.
Retroperitoneal angiosarcoma should be distinguished from the following pathological conditions: 1) pheochromocytoma (most patients with these tumors have a history of resistant hypertension, rapid blood pressure fluctuations, and are characterized by increased density, with a tendency for cystic degeneration and necrosis); 2) retroperitoneal lipoma (these masses are mainly ovoid or lobulated, soft, with smooth contours, surrounded by a capsule, and have distinctive features on ultrasound examination); 3) neuroblastoma, which is more common in children.
Surgical excision is the treatment of choice for angiosarcoma. However, the median survival of patients treated with surgery alone is 9 months, with 5-year survival around 20%. For patients who received adjuvant radiotherapy and chemotherapy, median survival can reach 36 months, while 5-year survival rises to 45%.The main causes of death are recurrence (local recurrence, incidence 75%) and distant metastases (incidence 34%). A multidisciplinary approach to these neoplasms is essential, but despite multimodal therapeutic management, the recurrence rate is high even in the case of small and localized tumors.
Conclusion
In conclusion, angiosarcoma is a rare, rapidly progressive, and highly aggressive tumor with a very poor prognosis, which can occur at any site in the body. Early diagnosis of retroperitoneal tumors is particularly difficult.A combination of clinical examination and imaging studies (ultrasound, CT scan, MRI, and PET scan) is required in order to draw safe conclusions about the nature of these retroperitoneal tumors. Percutaneous biopsy is extremely important, as it may contribute to early diagnosis.In order to achieve prolongation of survival in these patients, timely surgical excision of the neoplasm combined with targeted radiotherapy and chemotherapy is required.
- Retroperitoneal angiosarcoma is exceedingly rare with poor prognosis (median survival 12-16 months)
- IHC confirms endothelial origin: CD31+, CD34+, Factor VIII+
- Complete surgical resection with negative margins is the most important prognostic factor
- Paclitaxel-based chemotherapy and radiation are adjuvant options
- High recurrence rate necessitates close surveillance
Sign in to join the discussion
Loading...