Primary Angiosarcoma of the Spleen presenting as Idiopathic Thrombocytopenic Purpura.
Πρωτοπαθές Αγγειοσάρκωμα Σπληνός.
Introduction
Primary splenic malignancies are categorized into vascular and lymphoid, with the former originating from the red pulp and the latter from the white pulp. Splenic angiosarcoma is a rare and aggressive neoplasm originating from the vascular endothelium.The diagnosis and management of primary splenic angiosarcoma are usually delayed due to the varied manifestations and rarity of the disease. These malignant vascular splenic neoplasms may be misdiagnosed as benign vascular tumors or other non-vascular tumors due to the great variability in histological appearance.
We present the case of a 68-year-old female patient with a history of symptomatic anemia and thrombocytopenia who presented with massive splenomegaly due to primary splenic angiosarcoma.
Case Report
A 68-year-old female patient presented to our Hospital complaining of headaches, easy fatigue, chest pain, difficulty breathing/shortness of breath, and exertional dyspnea over the past 2 months. For these symptoms, she had been investigated at other hospitals with brain MRI, chest CT scan, bone marrow aspiration, and bone marrow biopsy.
No CNS pathology was identified. Hematological tests were indicative of peripheral-type thrombocytopenia, and despite thorough investigation, no serious cause for her hypersplenism was found.
On repeated questioning regarding her symptoms, she admitted the presence of diffuse left upper quadrant abdominal pain and unexplained loss of 8 kilograms of body weight.
Her personal medical history was unremarkable, apart from anemia and mild hypothyroidism.The patient disclosed that she suffered from long-standing, symptomatic, normochromic anemia and was being followed by a gastroenterologist to determine the probable etiology. She had previously been investigated multiple times with gastroscopies and colonoscopies without a diagnosis being established for her anemia.
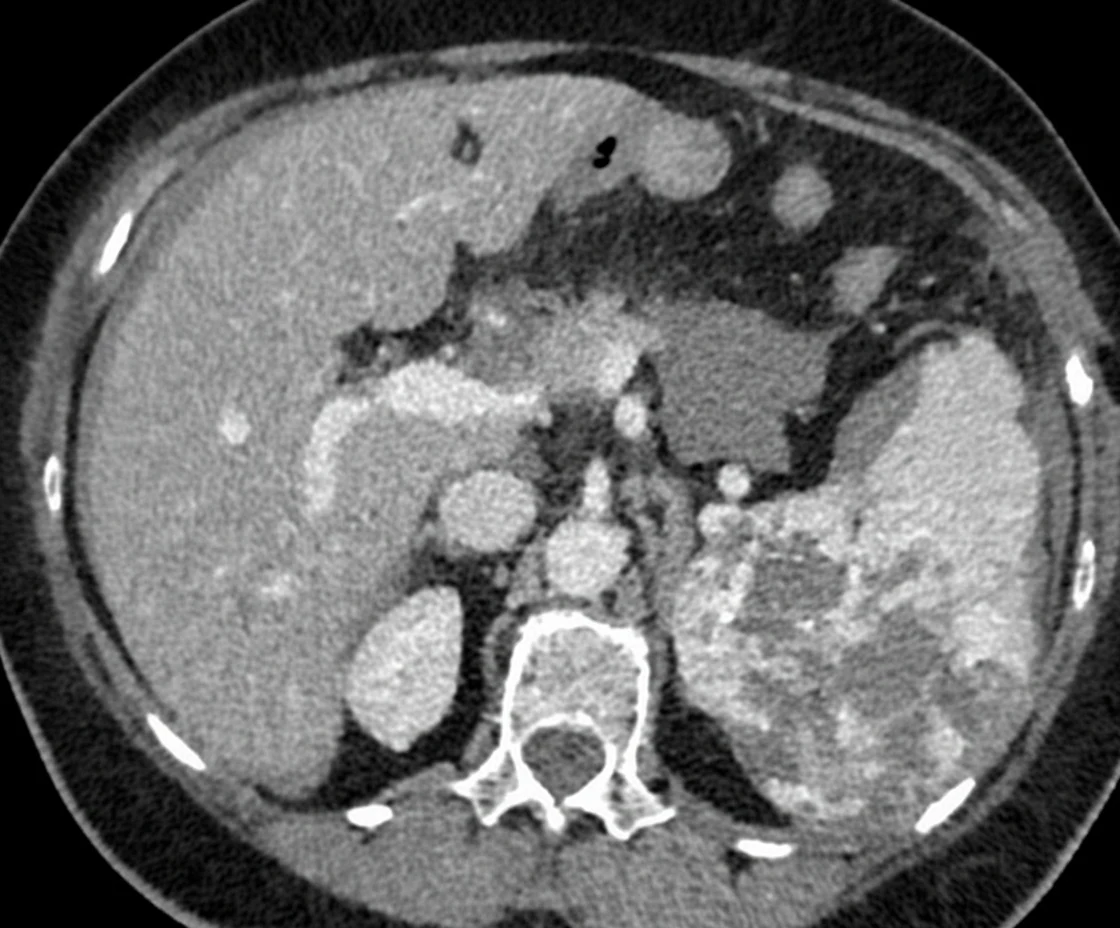
In the past, she received many packed red blood cell transfusions purely for symptomatic relief, since a reliable diagnosis could not be established. Laboratory workup revealed: white blood cell count 15.7 × 10⁹, platelet count 48 × 10⁹/L, and MCV 80.2.Abdominal CT revealed marked splenomegaly with the presence of multiple areas of low density.
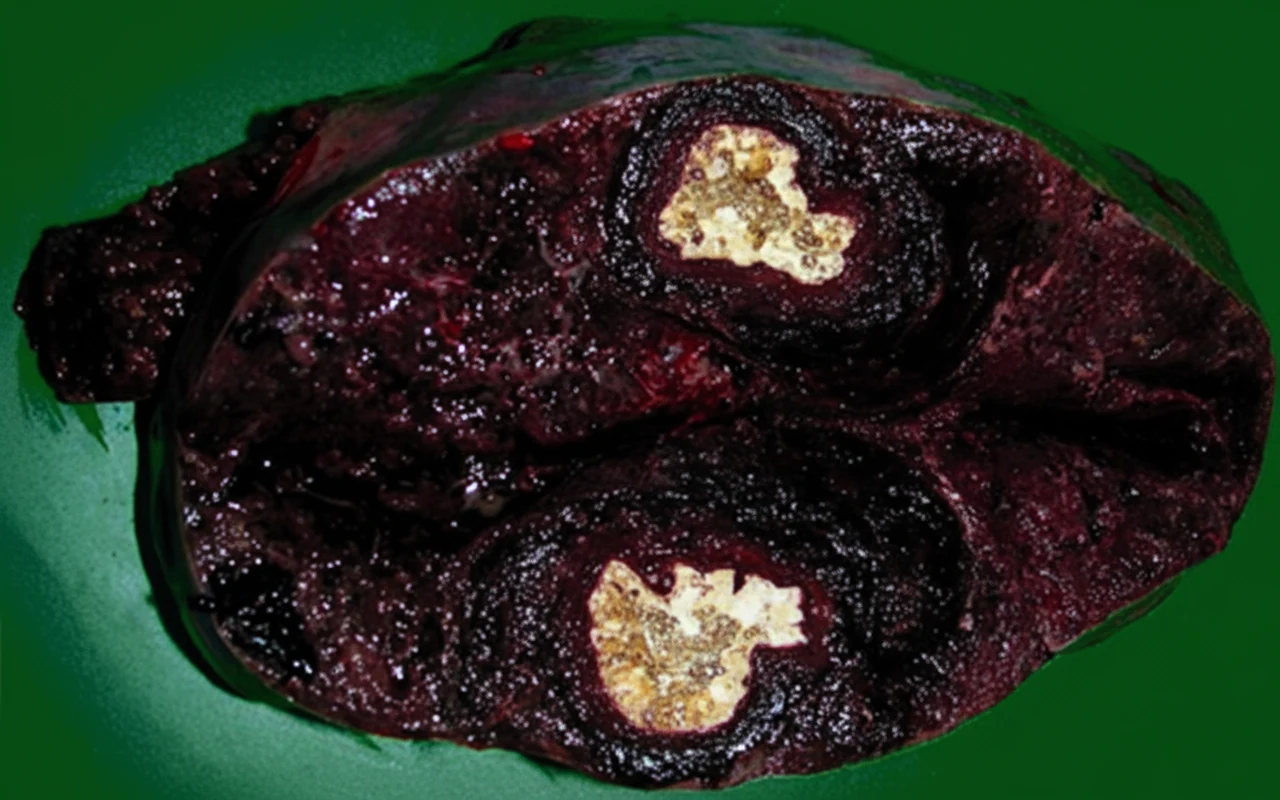
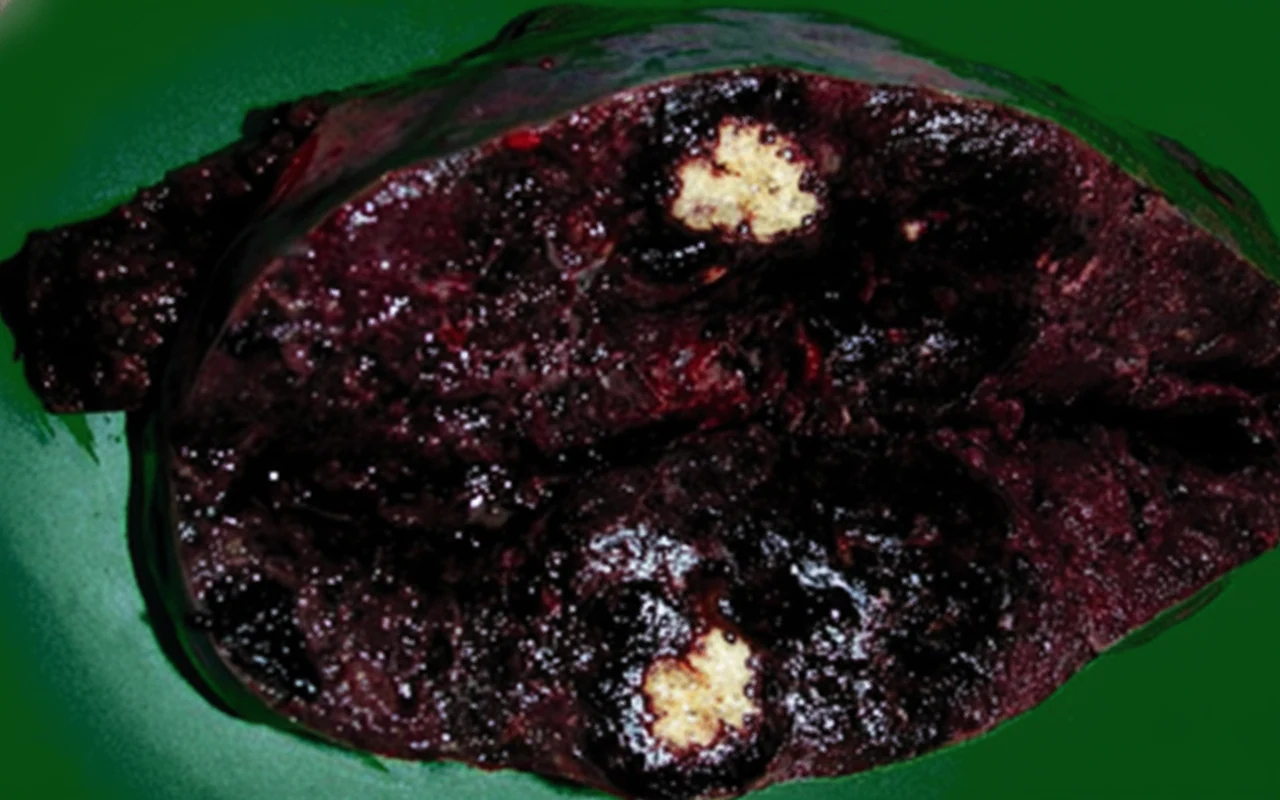
Due to the persistence of the patient's symptoms and the imaging findings, we decided to perform an elective diagnostic splenectomy.Upon entering the abdomen, we identified a large spleen with multiple adhesions to the omentum. Due to the organ's size, increased vascularity, and close contact with adjacent organs, we proceeded very carefully to remove the organ without rupture while simultaneously avoiding injury to neighboring organs such as the tail of the pancreas and stomach.The spleen was removed intact and weighed 972 grams.
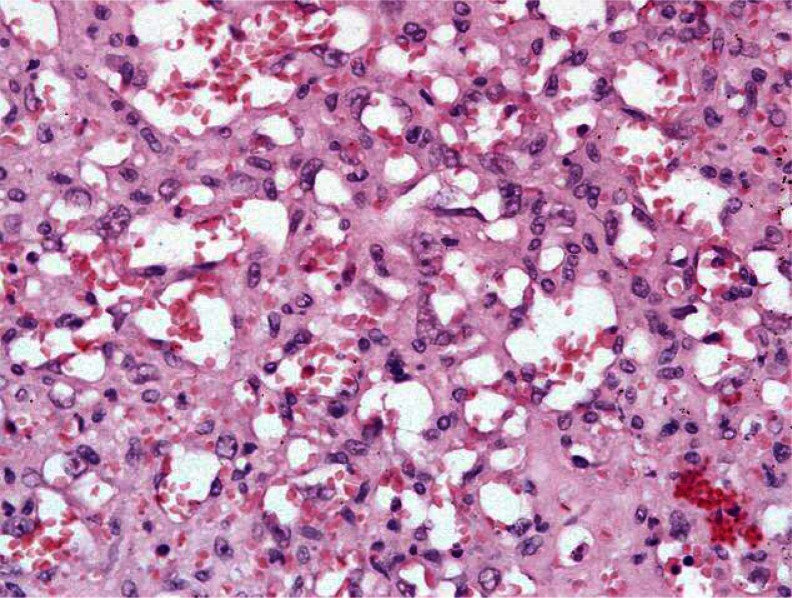
Histopathological examination revealed the presence of sheets and clusters of large atypical cells with vesicular chromatin and prominent nucleoli on a background of extensive necrosis, characteristic lymphocyte depletion, and the presence of abundant macrophages.The histological differential diagnosis was wide and included: 1) large cell lymphoma (B or T cell), 2) metastatic carcinoma, 3) melanoma, and 4) vascular neoplasms such as angiosarcoma and Kaposi sarcoma. Immunohistochemical examination identified that the large, atypical cells were of vascular origin, as they showed positivity for the vascular markers CD31, CD34, and von Willebrand factor.The Ki-67 index was 5–10%. Neoplastic cells were negative for numerous hematolymphoid markers such as CD3, CD20, CD8, CD45, CD61, CD138, lysozyme, and myeloperoxidase. Epithelial markers (cytokeratin, AE1/AE3) and S-100 were negative.
Therefore, the final diagnosis was splenic angiosarcoma and the patient was referred for oncological evaluation and further treatment.
Discussion
Angiosarcoma is a malignant neoplasm originating from vascular endothelial cells. These rare neoplasms constitute 2% of all soft tissue sarcomas and have shown a steady increase in incidence over the past 30 years.Angiosarcoma appears most frequently in the skin and superficial soft tissues but may also appear in other organs such as the breast, liver, spleen, and bones. Primary splenic angiosarcoma is extremely rare, with an annual incidence of 0.14–0.25 cases per million population.Splenic angiosarcoma is considered the most common non-lymphoid and non-hematopoietic malignant tumor of the spleen. Unlike angiosarcomas found in other organs, there is no association with occupational exposure to chemicals such as vinyl chloride, arsenic, or thorium dioxide.Although the malignancy can develop at any age, it occurs most commonly during the 5th and 6th decades of life, without sex or racial predilection.
The gross and microscopic characteristics of primary splenic angiosarcoma vary significantly, and therefore diagnosis is often challenging.Gross examination of the organ after splenectomy reveals a spleen weighing 250–3,200 grams, with massive splenomegaly defined as weight equal to or greater than 1,000 grams. In our case, the splenic weight was 972 grams and it occupied a large portion of the upper abdomen. Malignant vascular tumors of the spleen may be mistaken for benign vascular or malignant non-vascular tumors due to the variability of the histological picture.Although there are several different malignant morphological subtypes, the vasoformative pattern is the most common, more so than the poorly differentiated solid sarcomatous pattern. The overwhelming majority demonstrates a spongy or honeycomb-like proliferation involving an irregular anastomosing network of clefts or capillary-like spaces containing red blood cells.Frequently, these spongy areas may be combined with cavernous cystic spaces resembling cavernous hemangiomas with papillary sheets of proliferating neoplastic cells projecting into vascular lumina. While these architectural patterns are similar to those found in other benign vascular neoplasms, the distinguishing feature of angiosarcoma is the cytological atypia of the endothelial cell lining, which provides clear evidence of malignancy.
Neuhauser et al. describes additional features that help distinguish from benign vascular tumors, such as foci of necrosis, hemophagocytosis, the presence of hyaline globules, and extramedullary hematopoiesis. These neoplastic masses consist of malignant spindle-shaped, polygonal, epithelioid primitive, round, and multinucleated giant neoplastic cells.
Our patient's case was ultimately diagnosed as splenic angiosarcoma that appeared to have a vasoformative pattern containing large areas of cellular atypia with vesicular chromatin and prominent nucleoli. These malignant atypical cells also showed capillary-like channels.
The most sensitive and specific method for determining the endothelial origin of atypical cells is immunohistochemistry, as biopsy is dangerous and contraindicated to avoid organ rupture.
The differential diagnosis of splenic angiosarcoma includes other vascular tumors such as hemangiomas, littoral cell angiomas, lymphangiomas, hemangiopericytomas, and epithelioid vascular tumors.
A panel of immunohistochemical studies including markers of vascular and histiocytic differentiation can be quite helpful, confirming both the vascular origin of the tumor and histiocytic differentiation, which is unique to primary splenic angiosarcoma.The vast majority of these neoplasms demonstrate positivity for at least two markers of vascular differentiation, such as CD31, CD34, VEGF3, or von Willebrand factor (vWF). According to Kutok and Fletcher, CD31 is the most sensitive and specific marker, appearing positive even in the most poorly differentiated tumors, further confirming the diagnosis.Although long-term survivors are rare, no particular histopathological features have been identified that predict a more favorable outcome.
Diagnosis is a difficult challenge due to the non-specific symptoms and the rarity of the disease.This unfortunately entails diagnosis at an advanced stage. The spectrum of this disease is extremely variable, as in some cases it resembles vascular tumors such as hemangiomas, epithelioid hemangioendotheliomas, littoral cell angiomas, and Kaposi sarcomas, or malignant non-vascular tumors such as lymphangiomas, lymphomas, sarcomas, and metastatic carcinomas.
Imaging studies add identification features and contribute to diagnosis. Ultrasonography reveals multiple complex heterogeneous splenic masses, while splenomegaly is the most common finding, which however occurs in many pathological conditions.Contrast-enhanced CT reveals multiple hypervascular splenic masses. These lesions appear as round or irregular areas of low density, reflecting the presence of cystic or necrotic zones. However, lymphomas and metastatic disease frequently affect the spleen and can effectively mimic avascular angiosarcoma.
According to Falk et al., the most common presenting symptom is abdominal pain located in the left upper quadrant. Common systemic symptoms include weight loss, fatigue, and fever. Laboratory abnormalities include anemia, thrombocytopenia, and pancytopenia. The most common form of anemia accompanying splenic angiosarcoma is normocytic normochromic, with microcytic normochromic anemia being less frequent.With the onset of thrombocytopenia, the differential diagnosis expands to include various causes of immune platelet destruction, hypersplenism, and platelet production disorders. With the increase in splenic mass and vascularity, organ rupture may occur at rates reaching 13–32% of cases.This complication may lead to peritoneal dissemination with direct neoplastic tissue implants, hematogenous dissemination, or even fatal hemorrhage.
Our patient did not experience splenic rupture but exhibited chronic normocytic normochromic anemia, thrombocytopenia, weight loss, fatigue, and left upper quadrant abdominal pain.
The prognosis of splenic angiosarcoma continues to be poor. The mean survival of patients diagnosed with angiosarcoma is only 5 to 6 months if left untreated. Metastases are very common and generally appear rapidly, with the most common site being the liver, followed by the lungs, lymph nodes, and bones with decreasing frequency.Death occurs due to disease dissemination, or in untreated cases, due to splenic rupture resulting in hypovolemic shock and disseminated intravascular coagulation syndrome.
Primary angiosarcomas arising from visceral organs are extremely rare, and there are currently no established chemotherapy protocols.For the management of soft tissue sarcomas, doxorubicin and ifosfamide are used as single agents demonstrating response rates ranging between 16–36%.
In our patient's case, after splenectomy, she agreed to receive adjuvant chemotherapy consisting of doxorubicin and ifosfamide.The multimodal approach to her treatment helped keep her alive for at least 18 months after the initial diagnosis.
Conclusions
Primary splenic angiosarcoma constitutes a rare and aggressive neoplasm with a dismal prognosis. Its diagnosis and treatment pose a great challenge. While angiosarcomas of other organs are frequently associated with occupational exposure to vinyl chloride, arsenic, or the use of thorium dioxide contrast agent, this association does not exist for the spleen.Although there are several symptoms associated with this malignancy, none is specific for splenic angiosarcoma. We should suspect the presence of splenic angiosarcoma in patients complaining of persistent left upper quadrant abdominal pain with splenomegaly, systemic symptoms of malignancy, and persistent hematological abnormalities.
Imaging studies constitute a useful tool for determining the size and metabolic activity of the spleen, but alone are insufficient to establish the diagnosis.
The definitive diagnosis is established by histopathological examination and immunohistochemical staining.
The currently generally accepted therapeutic approach is splenectomy. Although there are some studies referring to adjuvant chemotherapy and radiotherapy, they do not constitute established specific therapies for the management of splenic angiosarcoma.Early diagnosis and surgical intervention remain of paramount importance.
- Primary splenic angiosarcoma is exceedingly rare with median survival of 5-6 months
- It may present as ITP, hemolytic anemia, or DIC without obvious tumor on imaging
- Diagnosis is often made incidentally on histopathology after splenectomy for presumed benign indications
- IHC markers: CD31+, CD34+, Factor VIII+ confirm endothelial origin
- Splenic angiosarcoma should be considered in the differential of splenomegaly with cytopenias
Sign in to join the discussion
Loading...